Cellular Allograft Products: Regulation, Manufacturing, and Safety

As bone graft materials and surgical techniques have evolved, there has been increasing interest in the use of stem cells for bone regeneration. Stem cells are unique cells in our bodies that can morph into a variety of different cells, including bone cells, and can improve the bone healing response.
Stem cells or other tissues used in bone grafting surgery come from two sources:
- Autologous Tissue (autograft) – stem cells or bone harvested from the patient being treated, or
- Allogeneic Tissue (allograft) – stem cells or bone taken from a deceased donor.
In the past 10 years, allograft stem cell products, also known as cellular allografts or allogeneic stem cell grafts, have become a popular bone grafting choice. Due to their source and composition, cellular allografts must adhere to certain protocols, regulations, and processes to ensure the donated tissue is safe to implant into another patient, and that it will be effective. This blog explains how these products are regulated, manufactured, and assessed for safety and efficacy.
For an overview of stem cell use in bone grafting, read more here.
Regulation of Cellular Allograft Bone Grafts
Typical cellular allografts in the bone graft market are classified by the FDA as “Human Cellular and Tissue-Based Products” (HCT/Ps). Due to this classification, they are not required to go through a regulatory clearance process that evaluates the product for safety or efficacy.
The reason behind this FDA classification is historical. When the FDA first began requiring medical devices to undergo a pre-market review for safety and efficacy in the 1970s, allogeneic tissue and organs were exempt because they were primarily processed by hospitals or regional, not-for-profit (NPO) tissue banks. Additionally, these were 100% donated tissue products with no additives and were, therefore, considered to be safe materials. At the time, there were no commercial organizations processing or distributing allografts. The processing was also very limited, primarily consisting of cutting, freeze-drying, or deep freezing the tissue. Even more extensive processes, like bone demineralization, were and still are considered by the FDA to be “minimally manipulated” and outside of its purview.
Today, although the processing of allogeneic tissue grafts for bone reconstruction has become significantly more complex, the FDA still deems it to be within the definition of “minimally manipulated.” Therefore, tissue grafts are referred to as “361 products” which implies no requirement for pre-market review of safety and efficacy. Conversely, any tissue grafts that are not within the guidelines for 361 are termed “351 products” and must receive biologic license application (BLA) approval by the FDA. This regulatory clearance process requires a detailed review of the product and must show clinical efficacy in a clinical trial before it can be distributed. Currently, there are no 351 tissue grafts for bone grafting approved for distribution in the US. A summary of 361 and 351 products is shown in Figure 1.
To qualify for the 361 HCT/P category, the FDA has two main requirements.
- The first requirement, termed “homologous use,” means that the tissue graft must be used in the recipient to regenerate tissue that is identical to the source of the tissue graft. For example, bone can be used as a bone graft while fat derived tissue cannot be used to regenerate bone.
- The second requirement is “minimal manipulation.” This refers to the use of minimal processing to manufacture the tissue product. Typical processes that are allowed include solution rinsing, freezing, freeze-drying, bone demineralization (where bone is soaked in acid to remove the mineral component), and sterilization methods. Specific to cellular allograft products, cells can be isolated from a donor using minimal manipulation techniques, but they cannot be cultured to increase cell number or exposed to solutions that would alter their cell type.

Manufacturing of Cellular Allograft Bone Grafts
Cellular allograft products are manufactured from human bones harvested from deceased donors. In the United States, the recovery of human organs and tissues is performed by tissue banks that, by law, are required to be not-for-profit organizations. These NPOs are responsible for collecting the tissues from hospitals shortly after a donor’s death, performing donor screening, processing the tissue into surgical products, and, in some organizations, distributing products to the hospitals. Aside from NPOs, the processing and distribution of allogeneic tissue can also be performed by a commercial, for-profit entity. Precisely how the cells for each commercial product are obtained is variable and is often associated with either patents or trade secrets. Consequently, it is difficult to know exactly how each commercial live tissue allograft product is manufactured.
Although the FDA does not review or approve cellular 361 allograft products before they enter the market, they do require all HCT/P products to be tested for certain bacteria, fungi, and viruses, and to be manufactured by Good Manufacturing Processes (GMP) quality standards. The facilities that process tissue allografts in the US are inspected by the FDA, and, in certain states, a state-based regulatory group. Additionally, most facilities are certified and inspected by voluntary organizations such as the American Association for Tissue Banks (AATB). Due to higher level of requirements by the AATB, the vast majority of hospitals and physicians require tissue products to be produced by an organization certified by AATB.
In cellular allograft products, the tissue consists of a liquid suspension of stem cells combined with a scaffold such as cortical or cancellous bone chips and/or demineralized bone matrix (DBM). DBM is a common choice since the acid solubilization of bone mineral results in a particulate tissue that is rich in osteoinductive growth factors. During cellular allograft manufacturing, the tissue is processed to ensure live cells are preserved. Due to the presence of live cells, the final product cannot be terminally sterilized, since this would kill the cells in addition to any biological contamination. Instead, cellular allograft products must be aseptically processed to preserve cell viability. Following processing, select units are then tested for sterility following AATB and FDA regulations before the lot can be released.
In addition to aseptic processing, cellular allograft manufacturing has additional requirements. In order to preserve the cellular component, bones from a single donor must be harvested and completely processed within 72 hours of donor death to maintain cell viability. This typically involves the long bones such as the femur, tibia, fibula, humerus, radius and ulna. Following harvesting, these bones are processed to isolate the stem cell component. Since the FDA and AATB require the use of a single donor per product lot, the bones are also processed into the other components of the product like cortical chips, cancellous chips, and/or DBM.
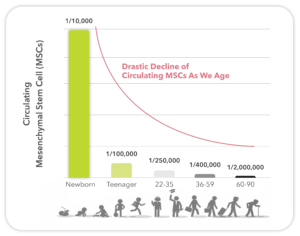
In addition to donor harvesting time, donor age is also an important consideration. Since the percentage of mesenchymal stem cells (MSCs) declines rapidly with age (Figure 2), donor selection based on age is critical. Typically, the “live cell” component is obtained from cancellous bones of the donor. The live cells are entrapped within the stroma and marrow of the cancellous bone. Consequently, the immunogenic mature cells, including blood cells, are removed as much as possible. This process requires extraction with inert tissue solvents and some physical separations methods.
Once all the tissues have been isolated and processed from a single donor, they are recombined to form the cellular allograft product. Since cellular allograft products must be stored frozen at low temperatures (< -70°C), a cryopreservative solution is added to maintain cell viability.
At this point, additional lot testing is conducted. This includes testing each lot for cell number, usually by hemocytometry, and viability, usually by trypan blue exclusion. In some products, specific antigens are stained and counted to ensure the cells are MSCs. This ensures that the product being sold has an acceptable number of nucleated cells, and that the cells are alive. However, the methods used to determine cell numbers are highly variable across manufacturers and are seldom reported. Specifications for cell viability and cell number at the time of packaging are reported, nominally at more than 70% and approximately one million cells per milliliter (ml or cc). Although manufacturers provide cell concentration data, these numbers can be misleading since a consistent counting method is not used between manufacturers and different cells can be included in the counts.
Following processing, testing, and lot release of a cellular allograft, the product is frozen and distributed to hospitals. After the products arrive at the hospital, they must be stored on-site in a low temperature freezer until used. Once it is time for use in the operating room (OR), the following process is typically performed:
- The cellular allograft unit is removed from the freezer and transported to the OR. The unit is removed from its packaging and warmed to room temperature.
- Once the tissue is thawed, the cryopreservative is quickly removed and replaced with saline.
- Due to the live cell component, the thawed unit must be implanted within 2 to 4 hours; otherwise, it must be disposed.
Safety of Cellular Allograft Products
As previously mentioned, cellular allograft products cannot be terminally sterilized without killing the cells, so each step must be carefully processed using aseptic technique. This involves processing the tissue using sterilized equipment and supplies and cleanroom processing. While the end product is tested for sterility, this testing only occurs on a small number of samples and units released for distribution are not tested.
Further, since the tissue is gently processed to maintain viable cells, other biological contaminants from the donor could also survive (e.g., live bacteria and viruses). As a result of this risk, the AATB requires extensive review of the donor’s medical and social history to exclude high risk patients. This includes an extensive list of common and uncommon bacteria, fungi, viruses, and prions. Additionally, the AATB has always prohibited combining tissues from different donors. This ensures that each tissue product is traceable to a particular patient-recipient and particular donor. That is why the live cells processed in allogeneic tissues are recombined with bone from the exact same donor.
Due to the donor pre-screening process and single donor per lot requirement, the incidence of disease transmission is rare. However, it is virtually impossible to test for every conceivable pathogen, and, occasionally, unfortunate incidents do occur. In 2021, one lot of allogeneic stem cells was processed from a donor that had a tuberculosis infection.1 The infection was present in the bone tissue and resulted in contaminated cellular allograft product being implanted in over 100 patients and causing multiple deaths. The FDA and AATB subsequently added testing for mycobacteria to their list of required micro-organisms.
The Future of Cellular Allografts in Bone Grafting
Cellular allograft bone graft products have been a popular graft choice for years. Surgeons are drawn to the live cellular component in the graft. Although stem cells can also be obtained from the patient’s own bone marrow aspirate, some surgeons like that stem cell allograft products do not require this secondary procedure. However, despite their popularity, cellular allografts have come under increased scrutiny by the FDA which has been magnified by the recent disease transmission event. Coupled with difficult storage conditions at hospitals and relatively high prices, some hospitals are now eliminating the use of cellular allograft at their facilities.
As such, BMA harvesting from the patient is seeing a resurgence in use. The use of patient-derived stem cells solves the issues associated with cellular allograft. Advanced bone graft systems such as Biogennix’s DirectCell® System provide the surgeon with all the necessary tools to create biologically active bone grafts with live, patient-derived stem cells in a safe manner that has no risk of disease transmission.
References:
- Schwartz, Noah G et al. “Nationwide tuberculosis outbreak in the USA linked to a bone graft product: an outbreak report.” The Lancet. Infectious diseases vol. 22,11 (2022): 1617-1625.
https://pubmed.ncbi.nlm.nih.gov/35934016/
Helpful links:
- YouTube: Overview of FDA 351 vs 361 Classifications on Biologics and HCTPs.
- The Lancet: Nationwide Tuberculosis Outbreak in the USA Linked To a Bone Graft Product.
- Becker’s Spine: More Lawsuits Filed Against Spine Biologics Company After Patients Sickened With Tuberculosis.
- JD Supra: What is The Aziyo FiberCel Case All About?
As a recognized leader in advanced bone graft technologies, Biogennix is committed to bringing high-quality educational content to our field. This blog will cover technical topics ranging from basic bone graft science to advanced osteobiologic principles. We’ll also discuss market trends and industry challenges. We thank you for reading and invite you to learn more about us here.